High-quality single-cell transcriptomics from tissue sections reveals histology-associated heterogeneity of mouse follicles
【Summary】
A research group led by Professor Kazuki Kurimoto and Assistant Professor Hiroki Ikeda from the Department of Embryology at Nara Medical University has developed a highly sensitive method for quantitative single-cell transcriptome analysis, which enables the precise examination of cells extracted from tissue sections using laser capture microdissection (LCM), and named the method DRaqL (Direct RNA recovery and quenching for LCM). Moreover, this method allows quantification of exon-exon junctions of mRNA. In an application of DRaqL to mouse ovarian sections, the research group established a predictive model for oocyte transcriptomes based on their diameter. Remarkably, deviations from this predictive model revealed growth retardation in oocyte gene expression profiles. This study also unveiled gene expression differences in granulosa cells, dependent on their proximity to oocytes. This research represents the first comprehensive quantitative analysis of the relationship between transcriptomes and histology during folliculogenesis at the single-cell level. This research was conducted in collaboration with Kyoto University (The Center for iPS Cell Research and Application [CiRA]/ Institute for the Advanced Study of Human Biology [WPI-ASHBi]), and RIKEN (RIKEN Center for Advanced Intelligence Project [AIP]). The results were published online at Life Science Alliance journal on September 18th, 2023
【Research back ground】
Multicellular organisms, including humans, begin their development from a single fertilized egg. Therefore, the quality control of oocytes is paramount for the stable inheritance of life, although the underlying mechanisms remain enigmatic. Oocytes are enveloped by supportive cells known as granulosa cells, forming follicles. In mouse ovaries, oocytes undergo a substantial size increase, ranging from less than 20 μm to over 70 μm in diameter. Granulosa cells play a crucial role by providing various substances to nurture oocytes and support their growth. To unravel the intricate mechanisms governing oocyte formation, it is important to establish a connection between their histological characteristics, such as their spatial relationship with granulosa cells and their size, with the precise transcriptome data of both oocytes and granulosa cells.
In recent decades, numerous single-cell transcriptomic methods have emerged, offering profound insights into the intricate workings of life and cellular processes. Nonetheless, the majority of these techniques depend dissociation of cells from tissues, leading to the consequence of losing histological information.
To address these limitations, recent advancements in spatial transcriptomics techniques have been developed, enabling gene expression analysis on tissue sections while preserving positional information. These methods have significantly enhanced spatial resolution, achieving single-cell level or even higher resolution. However, it is important to note that spatial transcriptomics methods involve trade-offs when it comes to the quality of gene expression analysis at the single-cell level. These trade-offs encompass challenges like quantifying exon-exon junctions for mRNA isoform analysis, relatively lower sensitivity, and difficulties in achieving unbiased transcriptome analysis, often due to probe-based analytical designs. Moreover, many spatial transcriptomics techniques necessitate the mounting of tissue sections onto costly glass slides designed for transcriptome analysis. This can make it challenging to select the appropriate sections for gene expression analysis based on microscopic inspection of tissue morphology. Additionally, highly scalable spatial transcriptomic approaches, which analyze entire sections at once, can become highly expensive when dealing with a large number of tissue sections. Furthermore, re-using these sections for other purposes, such as extracting genomic DNAs or proteins, becomes challenging.
On the other hand, laser capture microdissection (LCM) or similar techniques are employed to precisely cut and collect cells (or cell fragments) from tissue sections, which are then lysed for subsequent gene expression analysis. This approach offers complementary characteristics to current spatial transcriptomics methods. In addition to the practical usability mentioned above, LCM-based transcriptome analysis has the potential, in principle, to quantify single-cell transcriptomes from tissue sections with the same precision as regular, conventional transcriptomic analyses. Given the widespread use of LCM-based transcriptomics, enhancing its precision and sensitivity is expected to significantly advance various medical and life science research endeavors.
However, the microscopic analysis of tissue morphology demands the staining of sections fixed with alcohol or formalin to prevent the elution of cell contents during the experimental processes for staining. Using conventional methods, the efficiency of RNA extraction from fixed tissue sections has not always been optimal. These challenges have affected the accuracy and sensitivity of single-cell transcriptome analysis from tissue sections.
【Research achievement】
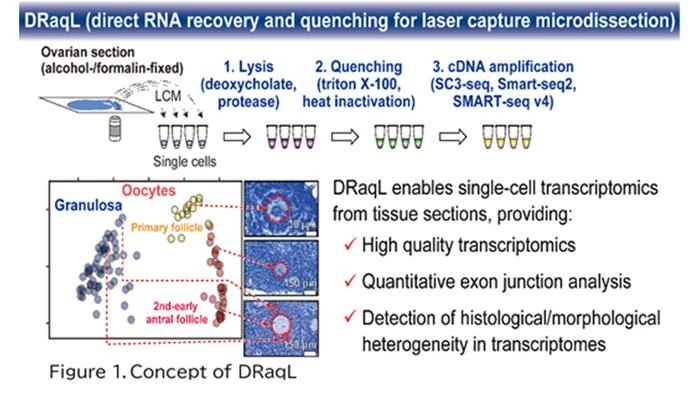
In this study, we have developed an efficient method for lysing cells (or cell fragments) isolated from alcohol- or formalin-fixed tissue sections and directly applying the lysate to RNA sequencing (RNA-seq). This technique involves treating the fixed tissue sections with two types of detergents: a denaturing detergent and a non-denaturing one. Initially, cells are efficiently lysed using the first denaturing detergent, sodium deoxycholate. Subsequently, a substantial quantity of the second non-denaturing detergent, Triton X-100, is added to counteract the denaturing effects of the initial detergent (Figure 1). This process facilitates both effective cell lysis and the enzymatic reactions required for subsequent RNA-seq. Moreover, when combined with protease treatment, even tissue sections rigidly fixed with formalin can be efficiently lysed. We named this lysis method DRaqL (direct RNA recovery and quenching for LCM) and have seamlessly integrated it with various cDNA amplification techniques, including the widely employed Smart-seq2, SMART-seq v4 (available as a kit), and SC3-seq (a method we have developed).
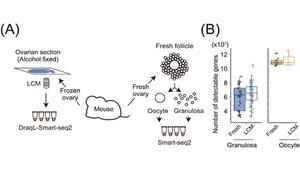
To assess the sensitivity of transcriptome analysis using DRaqL, we conducted RNA-seq on single cells isolated using LCM from mouse ovarian sections, as well as sections from cell blocks composed of homogeneous cultured cells. For comparison, we performed RNA-seq on freshly dissociated cells obtained from the same specimen (either another ovary from the same mouse or the same culture batch), without sectioning and fixation (Figure 2).
Comparing these transcriptome datasets revealed that single-cell RNA-seq from tissue sections exhibits sensitivity nearly identical to that of single-cell RNA-seq from freshly isolated cells. Moreover, the expression patterns were similar between the two methods. Additionally, we verified the quantitative capabilities of DRaqL-based single-cell RNA-seq for exon-exon junctions of mRNA.
Furthermore, the combination of DRaqL with protease treatment enables the analysis of single cells in tissue sections that have been strongly fixed with formalin. This analysis exhibits higher sensitivity than existing methods and demonstrates expression patterns akin to those of freshly isolated cells. These findings underscore the capacity of DRaqL to enable high-quality single-cell transcriptome analysis.
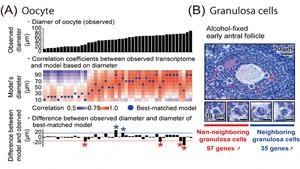
Furthermore, utilizing DRaqL, we explored the relationship between the morphology of mouse ovarian follicles and the transcriptome of their constituents, namely oocytes and granulosa cells (Figure 3). This investigation unveiled a continuous correlation between the size (diameter) of growing oocytes and their transcriptome, enabling the development of a predictive model for oocyte transcriptomes based on their size. Leveraging this model, we identified oocytes that deviated from the predicted patterns, displaying an immature transcriptome relative to their morphological growth. This finding may be associated with dominant follicle selection, a process wherein only dominant follicles progress during follicle development and culminate in ovulation.
In addition, with a focus on the spatial relationship between granulosa cells and oocytes, we conducted a comparative analysis between granulosa cells neighboring to oocytes and those distant from oocytes. This examination unveiled genes differentially expressed depending on the proximity to the oocytes, including regulators associated with transcription and cell proliferation.
The ability to perform quantitative single-cell transcriptomics in conjunction with ovarian follicle morphology relies on the amalgamation of precise microscopic examination of tissue sections with high-quality transcriptome analysis, accomplished in this study. In the future, the further development of transcriptomic analyses using DRaqL is anticipated to make significant contributions to fertility treatment and assisted reproductive medicine, including the identification of factors associated with the quality control of follicles and oocytes.
END