Heritable chronic cholestatic liver diseases
Chronic cholestasis, defined as the impairment of bile acid formation and/or flow persisting for more than six months, encompasses a broad spectrum of hepatobiliary disorders, both heritable and acquired. This review focuses on heritable causes of chronic cholestasis, which, although less common, present significant clinical challenges. Heritable chronic cholestatic liver diseases are typically diagnosed in childhood, but many cases present and persist into adulthood. This review aims to highlight the genetics, clinical pathophysiology, presentation, diagnosis, and treatment of these conditions.
Genetics and Pathophysiology
Heritable chronic cholestatic liver diseases result from genetic mutations affecting bile formation and transport. Key disorders include Congenital Hepatic Fibrosis (CHF), Caroli Disease (CD) and Caroli Syndrome (CS), Alagille Syndrome (ALGS), and Biliary Atresia (BA).
Congenital Hepatic Fibrosis (CHF)
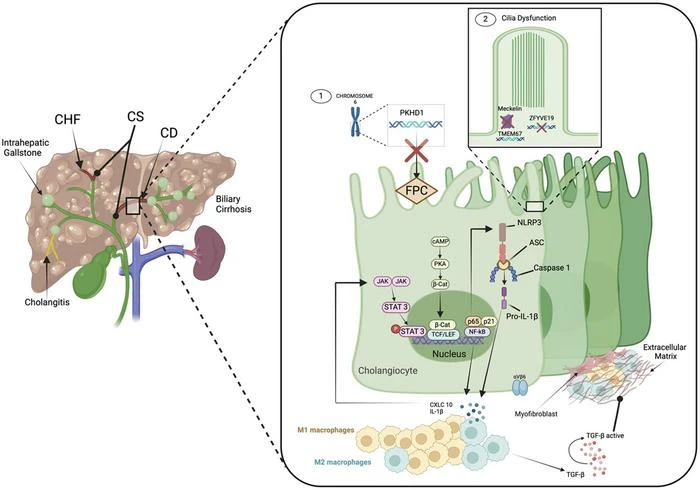
CHF is characterized by hepatic fibrosis and biliary tract changes, often presenting with portal hypertension and renal involvement. It is caused by mutations in the PKHD1 gene, leading to defective fibrocystin/polyductin protein. The incidence is approximately 1:20,000, with diagnosis typically occurring in early childhood. Clinical features include portal vein cavernous malformation, esophageal varices, and recurrent cholangitis. Management includes supportive care, endoscopic interventions, and liver transplantation (LT) in advanced cases.
Caroli Disease (CD) and Caroli Syndrome (CS)
CD and CS involve non-obstructive dilatation of the intrahepatic bile ducts, with CS also presenting with congenital hepatic fibrosis. Mutations in the PKHD1 gene are implicated, similar to CHF. Patients may experience recurrent cholangitis, liver abscesses, and portal hypertension. Treatment options include hemi-hepatectomy and liver transplantation.
Alagille Syndrome (ALGS)
ALGS is an autosomal dominant disorder affecting multiple organs, including the liver, heart, eyes, and bones. It is caused by mutations in the JAG1 or NOTCH2 genes, leading to bile duct paucity. Clinical manifestations include jaundice, pruritus, and cardiac defects. Management includes ursodeoxycholic acid (UDCA), fat-soluble vitamins, ileal bile acid transport (IBAT) inhibitors, and liver transplantation.
Biliary Atresia (BA)
BA is a progressive obliterative cholangiopathy affecting both intrahepatic and extrahepatic bile ducts, leading to fibrosis and cirrhosis. It is the most common indication for pediatric liver transplantation. Early diagnosis and surgical intervention, such as the Kasai portoenterostomy (KPE), are crucial. Long-term outcomes often necessitate liver transplantation due to progressive liver disease.
Clinical Presentation
Patients with heritable chronic cholestatic liver diseases often present with jaundice, pruritus, hepatomegaly, splenomegaly, and complications related to portal hypertension, such as variceal bleeding and ascites. Laboratory findings typically show elevated conjugated hyperbilirubinemia, alkaline phosphatase, and gamma-glutamyl transpeptidase.
Diagnosis
Diagnostic evaluation involves a combination of clinical assessment, laboratory tests, imaging studies, and genetic testing. Liver biopsy may be necessary to confirm the diagnosis and assess the extent of fibrosis. Genetic testing is particularly useful in identifying specific mutations and providing information for family counseling.
Management and Treatment
Management of heritable chronic cholestatic liver diseases focuses on symptom control, prevention of complications, and definitive treatment with liver transplantation when indicated. Supportive care includes nutritional support, management of pruritus, and treatment of portal hypertension. Specific therapies depend on the underlying genetic defect and disease severity.
Conclusion
Heritable chronic cholestatic liver diseases present significant clinical challenges due to their complex genetics and variable clinical presentation. Early diagnosis and appropriate management are essential to improve outcomes. Advances in genetic testing and targeted therapies hold promise for better management and understanding of these disorders. Future research should focus on elucidating the molecular mechanisms and developing novel therapeutic approaches to improve patient care and prognosis.
Full text
https://www.xiahepublishing.com/2310-8819/JCTH-2024-00119
The study was recently published in the Journal of Clinical and Translational Hepatology.
The Journal of Clinical and Translational Hepatology (JCTH) is owned by the Second Affiliated Hospital of Chongqing Medical University and published by XIA & HE Publishing Inc. JCTH publishes high quality, peer reviewed studies in the translational and clinical human health sciences of liver diseases. JCTH has established high standards for publication of original research, which are characterized by a study’s novelty, quality, and ethical conduct in the scientific process as well as in the communication of the research findings. Each issue includes articles by leading authorities on topics in hepatology that are germane to the most current challenges in the field. Special features include reports on the latest advances in drug development and technology that are relevant to liver diseases. Regular features of JCTH also include editorials, correspondences and invited commentaries on rapidly progressing areas in hepatology. All articles published by JCTH, both solicited and unsolicited, must pass our rigorous peer review process.
Follow us on X: @xiahepublishing
Follow us on LinkedIn: Xia & He Publishing Inc.
END