Massive fragment screen points way to new SARS-CoV-2 inhibitors
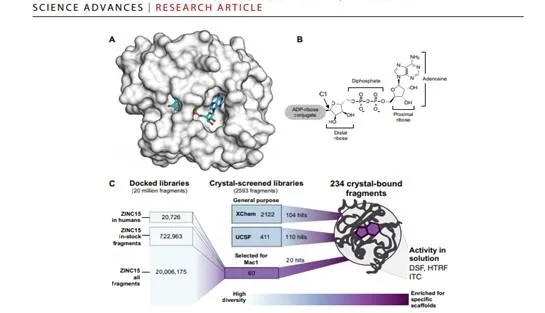
The study involved a crystallographic fragment screen of the Nsp3 Mac1 protein by an open science collaboration between researchers from the University of Oxford, the XChem platform at Diamond Light Source, the UK's national synchrotron, and researchers from the QCRG Structural Biology Consortium at the University of California San Francisco. The international effort discovered 234 fragment compounds that directly bind to sites of interest on the surface of the protein, and map out chemical motifs and protein-compound interactions that researchers and pharmaceutical companies can draw on to design compounds that could be developed into antiviral drugs. This work is thus foundational for preparing for future pandemics.
"Robustly identifying this kind of chemical matter for promising and tractable targets like Nsp3 is a first step in rational drug discovery. This is always a long journey fraught with difficulty and failure, but the battery of new structural biology methods that we combined in this study, including fragment screening at Diamond and computational docking at UCSF, are helping to change drug discovery and make it easier to find effective drug candidates," comments Principal Beamline Scientist, Frank von Delft.
These fragments cover a wide range of chemical motifs, and the study lays out the next steps of designing more elaborate molecules that combine the observed themes, synthesizing them and confirming experimentally whether they strongly bind the protein and have a biological effect. The most promising compounds can then be progressed in fully-fledged drug discovery programmes, which includes not only improving the biological potency but also ensuring the final molecule has important drug properties such as easy absorption and minimal side effects.
Most drugs contain a few key components that cause the desired, effect while the rest of the molecule may be important for other reasons, such as solubility, uptake from the gut or how the drug is processed by our metabolism. Traditional high-throughput screening entails testing very large collections of bigger, generally sub-optimal molecules, which are experiment of great complexity.
Instead, fragment screening is an approach for identifying building blocks of the future drug molecule, observing how they interact with the protein under study, contextualizing those interactions, and providing starting points for molecules that directly influence the biology of the protein. This method significantly reduces the number of compounds that need to be screened to find one that really binds, while still informing a broad range of potential molecules. Doing the experiment by structural biology, as implemented at the XChem platform, yields this information directly in 3D, greatly accelerating up the design process and ensuring a far more cost-effective overall experiment.
The UCSF collaborators also used another innovative drug discovery technique, Computational Docking. This deploys computer models and simulations to assess the likely interactions of virtual molecules for favourable interactions with Mac1 and their promise as starting points for drug discovery. The team identified 60 candidates from a virtual library of 20 million molecules, which were then experimentally tested using X-ray crystallography, yielding 20 good hits.
"This is a significantly higher-than-random hit rate, validating the new specific docking methodologies developed by our UCSF colleagues. The high quality structural data of Mac1 that we obtained by X-ray crystallography was essential, but the validation of the approach means that in future, we have additional power for exploring compounds that are not physically available. Overall, this work not only accelerates our ability to validate whether targeting NSP3 Mac1 is an effective way to develop antivirals; it also is hugely valuable in improving the template of methodologies for future inhibitor discovery and development throughout the community of drug discovery," concludes Frank von Delft.
INFORMATION:
For further information please contact Diamond Communications: Lorna Campbell +44 7836 625999 or Isabelle Boscaro-Clarke +44 1235 778130
Science Advances Paper: Fragment binding to the Nsp3 macrodomain of SARS-CoV-2 identified through crystallographic screening and computational docking advances.sciencemag.org/cgi/content/full/7/16/eabf8711/DC1
Published 14 April 2021 10.1126/sciadv.abf8711
Summary: The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) macrodomain within the nonstructural protein 3 counteracts host-mediated antiviral adenosine diphosphate ribosylation signalling. This enzyme is a promising antiviral target because catalytic mutations render viruses nonpathogenic. Here, we report a massive crystallographic screening and computational docking effort, identifying new chemical matter primarily targeting the active site of the macrodomain. Crystallographic screening of diverse fragment libraries resulted in 214 unique macrodomain-binding fragments of 2533 screened. An additional 60 molecules were selected from docking more than 20 million fragments, of which 20 were crystallographically confirmed. X-ray data collection to ultra-high resolution and at physiological temperature enabled assessment of the conformational heterogeneity around the active site. Several crystallographic and docking fragment hits were also confirmed by solution binding using three biophysical techniques (differential scanning fluorimetry, homogeneous time-resolved fluorescence, and isothermal titration calorimetry). The 234 fragment structures presented explore a wide range of chemotypes and provide starting points for development of potent SARS-CoV-2 macrodomain inhibitors
Authors: Marion Schuller, Galen J. Corey, Stefan Gahbauer, Daren Fearon, Taiasean Wu, Roberto Efraín Díaz, Iris D. Young, Luan Carvalho Martins, Dominique H. Smith, Ursula Schulze-Gahmen, Tristan W. Owens, Ishan Deshpande, Gregory E. Merz, Aye C. Thwin, Justin T. Biel, Jessica K. Peters, Michelle Moritz, Nadia Herrera, Huong T. Kratochvil, QCRG Structural Biology Consortium, Anthony Aimon, James M. Bennett, Jose Brandao Neto, Aina E. Cohen, Alexandre Dias, Alice Douangamath, Louise Dunnett, Oleg Fedorov, Matteo P. Ferla, Martin R. Fuchs, Tyler J. Gorrie-Stone, James M. Holton, Michael G. Johnson, Tobias Krojer, George Meigs, Ailsa J. Powell, Johannes Gregor Matthias Rack, Victor L. Rangel, Silvia Russi, Rachael E. Skyner, Clyde A. Smith, Alexei S. Soares, Jennifer L. Wierman, Kang Zhu, Peter O'Brien, Natalia Jura, Alan Ashworth, John J. Irwin, Michael C. Thompson, Jason E. Gestwicki, Frank von Delft, Brian K. Shoichet, James S. Fraser, Ivan Ahel
About Diamond Light Source: W: http://www.diamond.ac.uk Twitter: @DiamondLightSou
Diamond Light Source provides industrial and academic user communities with access to state-of-the-art analytical tools to enable world-changing science. Shaped like a huge ring, it works like a giant microscope, accelerating electrons to near light speeds, to produce a light 10 billion times brighter than the Sun, which is then directed off into 33 laboratories known as 'beamlines'. In addition to these, Diamond offers access to several integrated laboratories including the world-class Electron Bio-imaging Centre (eBIC) and the Electron Physical Science Imaging Centre (ePSIC).
Diamond serves as an agent of change, addressing 21st century challenges such as disease, clean energy, food security and more. Since operations started, more than 14,000 researchers from both academia and industry have used Diamond to conduct experiments, with the support of approximately 760 world-class staff. More than 10,000 scientific articles have been published by our users and scientists.
Funded by the UK Government through the Science and Technology Facilities Council (STFC), and by the Wellcome Trust, Diamond is one of the most advanced scientific facilities in the world, and its pioneering capabilities are helping to keep the UK at the forefront of scientific research.