Deep-learning framework advances tissue analysis in spatial transcriptomics
Biological tissues are made up of different cell types arranged in specific patterns, which are essential to their proper functioning. Understanding these spatial arrangements is important when studying how cells interact and respond to changes in their environment, as well as the intricacies of pathologies like cancer. Spatial transcriptomics (ST) techniques, which have been rapidly evolving over the past decade, allow scientists to map gene activity within tissues while keeping their structure intact, offering deeper insights into both healthy and diseased states.
However, identifying distinct tissue regions based on their gene activity remains a challenge, as different methods struggle to balance genetic data with spatial organization. For example, some ST methods rely on arbitrarily defined distance parameters, which may not accurately reflect biological boundaries. Others incorporate multiple tissue images to improve accuracy but are limited by inconsistencies in image quality and data availability. Additionally, comparing image data coming from different experiments can be difficult due to technical differences, often requiring manual adjustments to align datasets properly and achieve batch integration.
To address these problems, a research team led by Professor Kenta Nakai of the Institute of Medical Science, The University of Tokyo, Japan, developed a deep-learning framework called Spatial Transcriptomics Analysis via Image-Aided Graph Contrastive Learning (STAIG). This study, published online in Nature Communications on January 27, 2025, introduces the STAIG framework, which integrates gene expression, spatial data, and histological images without the need for manual alignment, yielding exceptional results. The study was co-authored by several people from Prof. Nakai’s lab, notably Yitao Yang, a PhD student under his supervision.
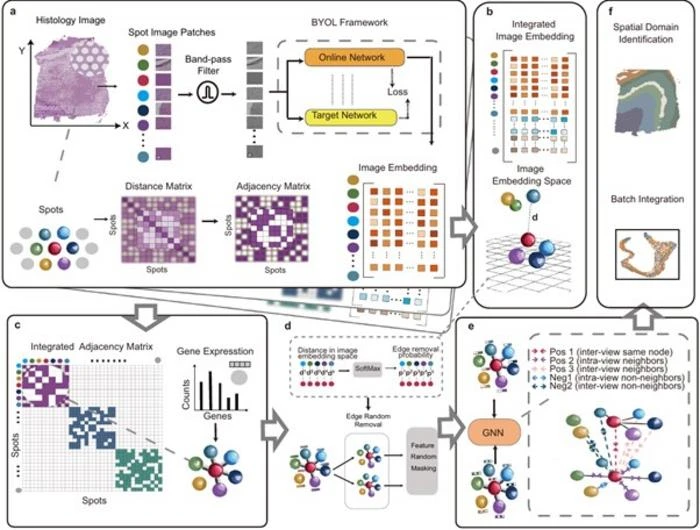
STAIG processes histological images by segmenting them into small patches and extracting features using a self-supervised model, eliminating the need for extensive pre-training. It then constructs a graph structure from these features, strategically integrating spatial information to effectively manage vertically stacked images. In these graphs, nodes represent gene expression data, while edges reflect spatial adjacency. Using an advanced approach called graph contrastive learning, STAIG identifies key spatial features, enabling it to map distinct gene expression patterns to specific tissue regions. “STAIG leverages a robust model architecture and additional image data to achieve high-accuracy spatial domain identification, while also enabling batch integration without the need to align tissue sections or perform manual adjustments,” says Nakai, outlining some of the main advantages of the model.
The research team conducted extensive benchmark evaluations, comparing STAIG to other state-of-the-art ST techniques. The results demonstrated STAIG’s superior performance across various conditions, including cases where spatial alignment was unavailable or histological images were missing. In datasets of human breast cancer and zebrafish melanoma, STAIG successfully identified spatial regions with high resolution, including challenging regions that existing methods struggled to detect. Additionally, it precisely delineated tumor boundaries and transitional zones, showcasing its potential for cancer research.
The researchers have high hopes for their proposed framework and its potential applications in medical research and biology. “STAIG will accelerate the use of spatial transcriptome data to understand the complex structures of biological systems, including the interaction between cancer cells and their surrounding cells and the formation of organs in developing embryos,” concludes Nakai, “Our study will enhance our understanding of how our brain works, how cancer cells develop, and how our body is constructed. Such knowledge will stimulate the development of new therapeutic methods for a variety of diseases.”
Further research in this field will let us fully harness the power of spatial transcriptomics!
***
Reference
Authors: Yitao Yang1, Yang Cui1, Xin Zeng1, Yubo Zhang1, Martin Loza2, Sung-Joon Park2 and Kenta Nakai1,2,
Title of original paper: STAIG: Spatial transcriptomics analysis via image-aided graph contrastive learning for domain exploration and alignment-free integration
Journal: Nature Communications
DOI: 10.1038/s41467-025-56276-0
Affiliations:
1Department of Computational Biology and Medical Science, Graduate School of Frontier Sciences, The University of Tokyo, Japan
2Human Genome Center, the Institute of Medical Science, The University of Tokyo, Japan
About The Institute of Medical Science, The University of Tokyo
The Institute of Medical Science, The University of Tokyo (IMSUT), established in 1892 as the Institute of Infectious Diseases and renamed IMSUT in 1967, is a leading research institution with a rich history spanning over 127 years. It focuses on exploring biological phenomena and disease principles to develop innovative strategies for disease prevention and treatment. IMSUT fosters a collaborative, interdisciplinary research environment and is known for its work in genomic medicine, regenerative medicine, and advanced medical approaches like gene therapy and AI in healthcare. It operates core research departments and numerous specialized centers, including the Human Genome Center and the Advanced Clinical Research Center, and is recognized as Japan’s only International Joint Usage/Research Center in life sciences.
About Professor Kenta Nakai from the Institute of Medical Science, The University of Tokyo
Kenta Nakai is a Professor at the Institute of Medical Science, the Department of Computer Science, Graduate School of Information Science and Technology, and the Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences at The University of Tokyo. He obtained his PhD from Kyoto University in 1992. He specializes in sequence analysis in molecular biology, bioinformatics, and genome analysis. He has published over 200 papers on these topics.
Funding information
This study was supported by the JSPS KAKENHI [22K06189 to K.N., JP22K21301 to M.L., and 20H05940 to S.P.] and the JST SPRING [JPMJSP2108 to X.Z.].
END