Ferroptosis in regulating treatment tolerance of digestive system tumors
The global burden of digestive tract tumors is profound, with these cancers accounting for nearly half of all malignant tumors worldwide. Despite advancements in endoscopic diagnostic methods, which enable earlier detection and treatment, a large portion of patients still receive diagnoses at later stages. For these patients, chemotherapy, radiation, and immunotherapies are often the only viable options, yet resistance to treatment remains common, leading to high recurrence and mortality rates. Research increasingly shows that ferroptosis may be a key mechanism to reverse treatment tolerance. Ferroptosis, characterized by iron-mediated lipid peroxidation, holds the potential to counter resistance by selectively targeting cancer cells’ defense mechanisms, paving the way for new therapeutic strategies.
Ferroptosis and Tumor Treatment Resistance
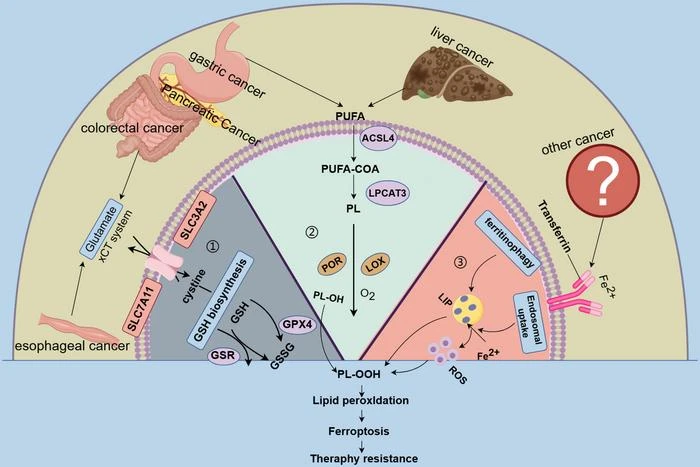
Ferroptosis represents a distinct cell death pathway regulated by iron metabolism, ROS production, and lipid peroxidation. The primary trigger for ferroptosis involves excessive accumulation of lipid peroxides within cell membranes, particularly in iron-rich environments. This peroxidation compromises membrane integrity, ultimately leading to cell death. Various molecular pathways, such as glutathione peroxidase 4 (GPX4) inhibition, iron metabolism deregulation, and polyunsaturated fatty acid metabolism, converge to regulate ferroptosis.
Tumor cells often activate endogenous stress relief mechanisms to protect themselves from oxidative damage and chemotherapy-induced stress. Persister cells (PCs) and cancer stem cells (CSCs), which exhibit heightened resistance to treatment, are two cell populations in which ferroptosis plays a particularly important role. PCs and CSCs can adapt to repeated chemotherapy and other treatments, becoming more resilient and challenging to eradicate. Inducing ferroptosis in these cell populations offers a promising pathway to decrease treatment resistance.
Ferroptosis in Specific Gastrointestinal Tumors
Gastric Cancer
Gastric cancer is often refractory to treatment due to resistance mechanisms that involve antioxidant pathways. ROS modulation and GPX4 downregulation appear to enhance sensitivity to chemotherapeutic agents like cisplatin. Studies have shown that peroxiredoxin-2, an enzyme that reduces cellular ROS levels, can increase sensitivity to cisplatin by promoting ferroptosis. Additionally, pathways involving general control nonderepressible 2 (GCN2), eIF2α, and ATF4 have been linked to gastric cancer’s adaptation to oxidative stress. Targeting these pathways to induce ferroptosis could help overcome cisplatin resistance. Sorafenib, a tyrosine kinase inhibitor, has also shown potential in gastric cancer by promoting ferroptosis through ATF2-related pathways, further supporting the potential for ferroptosis-based therapeutic strategies.
Colorectal Cancer
Colorectal cancer treatment is complicated by resistance to chemotherapy and targeted therapies, leading to poor prognoses in advanced cases. Lipid metabolism and iron regulation play crucial roles in regulating ferroptosis in colorectal cancer cells. For instance, the KIF20A-NUAK1-PP1β-GPX4 pathway mediates resistance to oxaliplatin by modulating ferroptosis. Studies indicate that cysteine desulfurase (NFS1) deficiency can synergize with oxaliplatin to promote ferroptosis, elevating ROS levels and improving chemosensitivity. Additionally, FAM98A has been found to increase tolerance to 5-fluorouracil by inhibiting ferroptosis. Resistance to therapies targeting the epidermal growth factor receptor (EGFR) pathway also appears to be reversible through ferroptosis. Combining cetuximab with β-elemene, a ferroptosis inducer, effectively sensitizes KRAS-mutant colorectal cancer cells to treatment, highlighting ferroptosis as a potential countermeasure to drug resistance.
Pancreatic Cancer
Pancreatic cancer is often diagnosed at an advanced stage, with only 20% of patients qualifying for surgical treatment. This aggressive cancer is generally treated with gemcitabine and other chemotherapies, though resistance to these agents is a frequent challenge. Ferroptosis inducers (FINS) have shown promise in enhancing sensitivity to chemotherapy by inducing cell death in resistant pancreatic cancer cells. Gemcitabine, in particular, can stimulate ROS accumulation and activate ferroptosis by upregulating p22-phox expression, leading to further ROS accumulation and cellular damage. Another key resistance mechanism involves the nuclear factor erythroid 2–related factor 2 (NRF2), which regulates antioxidant defenses. Inhibiting NRF2 can enhance the effects of ferroptosis inducers, offering a potential strategy to improve pancreatic cancer responses to chemotherapy.
Liver Cancer
For patients with liver cancer who are unsuitable for surgery, sorafenib, a multi-kinase inhibitor, remains a standard treatment. However, resistance to sorafenib can severely limit its efficacy. Ferroptosis inducers can enhance sorafenib sensitivity in liver cancer cells, offering a targeted approach to overcome resistance. MicroRNA miR-23a-3p has been identified as a negative regulator of ferroptosis, protecting hepatocellular carcinoma cells from lipid peroxidation and iron accumulation. Reducing miR-23a-3p levels increases responsiveness to sorafenib, as does downregulating metallothionein-1G (MT-1G), which inhibits ferroptosis. These pathways represent potential therapeutic targets to reverse treatment resistance.
Esophageal Cancer
Esophageal cancer, commonly treated with chemoradiotherapy and surgery, often develops resistance to treatment. Targeting ferroptosis-related pathways, such as system Xc-, GPX4, and NRF2, could improve sensitivity to therapies. Emerging immunotherapies, like PD-1 and PD-L1 inhibitors, have also been linked to ferroptosis induction. PD-L1 antibodies combined with ferroptosis inducers can promote tumor cell death through lipid peroxidation pathways. T-cell-mediated cytotoxicity also contributes to this process, suggesting that immunotherapy combined with ferroptosis inducers may offer new treatment avenues for esophageal cancer.
Limitations and Future Perspectives
While ferroptosis shows potential as a therapeutic target, research is still in the early stages, and several limitations must be addressed. Sensitivity to ferroptosis varies among tissues, and the combinatorial effects of ferroptosis inducers with conventional therapies remain unclear. Current detection techniques for ferroptosis, such as ROS and lipid peroxidation measurements, flow cytometry, and Western blotting, lack a single gold standard, though combining methods provides a more comprehensive assessment. Future research should focus on developing more precise ferroptosis detection tools, exploring biomarkers, and studying the antagonistic and synergistic effects of combined therapies.
Conclusions
Ferroptosis represents a promising approach to overcoming treatment resistance in digestive system tumors. The induction of ferroptosis through mechanisms involving GPX4 inhibition, ROS modulation, and lipid peroxidation offers a potential strategy to selectively target resistant tumor cells, particularly PCs and CSCs. Future clinical and basic research on ferroptosis could lead to more effective cancer treatments, reduce disease burden, and improve quality of life for patients with advanced digestive system cancers.
Full text
https://www.xiahepublishing.com/2835-3315/CSP-2024-00018
The study was recently published in the Cancer Screening and Prevention.
Cancer Screening and Prevention (CSP) publishes high-quality research and review articles related to cancer screening and prevention. It aims to provide a platform for studies that develop innovative and creative strategies and precise models for screening, early detection, and prevention of various cancers. Studies on the integration of precision cancer prevention multiomics where cancer screening, early detection and prevention regimens can precisely reflect the risk of cancer from dissected genomic and environmental parameters are particularly welcome.
Follow us on X: @xiahepublishing
Follow us on LinkedIn: Xia & He Publishing Inc.
END