TLE6 identified as a protein associated with infertility in male mice
Infertility is a major global challenge associated with physiological and psychological impact. Genetic mutations that affect early embryonic development, oocyte (egg cell) maturation, and fertilization have recently been studied as causes of infertility. One of the most well-studied causes of early embryonic infertility is mutations in the subcortical maternal complex (SCMC)-related genes.
SCMC participates in embryo development and cleavage by maintaining the structure of the egg cytoplasm and recruiting proteins that assist proper embryo formation. SCMC is composed of multiple proteins, of which the transducin-like enhancer of split 6 (TLE6) is the most crucial member. In the absence of TLE6, the structural integrity of SCMC is compromised, and consequently cell division in the embryo fails after the two-cell stage, resulting in embryo fragmentation and death. There is ample evidence supporting the role of TLE6 in female infertility, but its role in male germ cells remains unexplored.
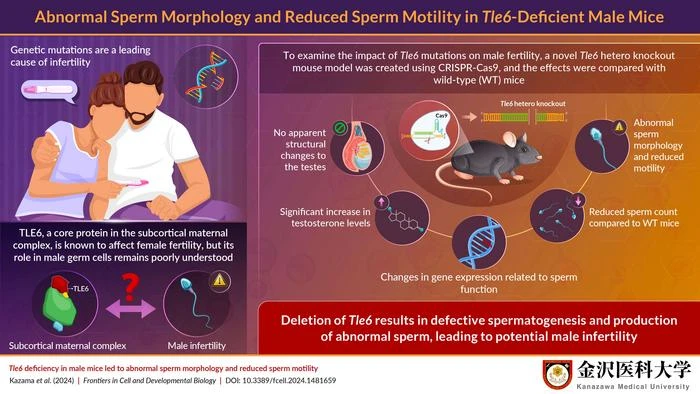
To address this gap, Mr. Kousuke Kazama, a Research Associate, from the Research Support Center, Medical Research Institute, Kanazawa Medical University, Japan, along with Dr. Hirofumi Nishizono and Ms. Yuki Miyagoshi, also from Kanazawa Medical University, attempted to understand the effects of Tle6 gene deficiency on male fertility using the Tle6 deficient mouse model. They developed a novel Tle6 gene hetero knockout male mouse model using a technique called CRISPR-Cas9 that enables the editing of genes. Their findings were published in Volume 12 of Frontiers in Cell and Developmental Biology on October 24, 2024.
“We generated Tle6 hetero knockout mice to investigate the effects of Tle6 deficiency in male mice. We performed genome editing of the embryos using the CRISPR-Cas9 system and electroporation to generate the Tle6 hetero knockout mice,” states Kazama, explaining the main methodology used in the study. To investigate whether Tle6 deficiency leads to erratic mating behavior, Tle6-deficient and wild-type (WT) male mice were mated with WT female mice. The mating frequency and the number of offspring did not differ between Tle6-deficient and WT mice. Additionally, embryos derived from the sperm of Tle6-deficient male mice showed similar developmental rates as those derived from WT male mouse sperm.
The question of why Tle6 deficiency-related traits were not transmitted to the next generation prompted the researchers to further explore the gene’s role in sperm function. Kazama elaborates, “We hypothesized that the difficulty in transmitting genetic traits from Tle6-deficient male mice could be due to reduced sperm count and motility.” To test this hypothesis, they analyzed the testes and sperm of Tle6-deficient male mice. While the structure of the testes was not affected due to Tle6 deficiency, they found a significant reduction in sperm count and a marked decrease in the number of motile sperm. Moreover, 57% of the sperm from Tle6-deficient mice had an abnormal head structure, and 7% were double-headed. The researchers suspected dysregulated hormone levels in these mice and consequently found elevated levels of testosterone (an important sex hormone) in Tle6 hetero knockout male mice.
Visualization of sperm from WT and Tle6 knockout mice using immunofluorescence staining revealed that TLE6 protein in deficient mice was localized in the sperm midpiece. This region overlapped with the location of mitochondria, which are important for energy production, suggesting that TLE6 might play a role in energy production in the sperm. Gene expression related to fertilization, sperm motility, and sperm structure in the testes of Tle6-deficient mice showed an overall increase.
Together, the findings of this study highlighted the impact of Tle6 deficiency in male mice and its role in potential male infertility. “The role of TLE6 in the development of sperm cells may vary between humans and mice. Therefore, further research is necessary to clarify the mechanisms by which Tle6 deficiency causes sperm abnormalities in Tle6 hetero knockout mice and to explore its clinical relevance in humans,” concludes Kazama.
In summary, this study sheds further light on male infertility and paves the way for more advanced research and the development of new assisted reproductive technologies.
***
Reference
Title of original paper: Tle6 deficiency in male mice led to abnormal sperm morphology and reduced sperm motility
Journal: Frontiers in Cell and Developmental Biology
DOI: https://doi.org/10.3389/fcell.2024.1481659
About Kanazawa Medical University (KMU), Japan
Kanazawa Medical University (KMU) is a reputed medical school located in Ishikawa, Japan. Established in 1972 with Albert Schweitzer's idea of “Ehrfurcht vor dem Leben (respect for life) and five “S” keywords: Spirit, Science, Skill, Speed, and Safety, KMU is pledged to the development of high-quality medical professionals. KMU continues to grow by engaging nearly 4,500 graduates from the School of Medicine in medical care globally. Furthermore, KMU sends over 2,500 nurses, public health nurses, and midwives to gain practical exposure in the medical field. By following the motto and five “S” elements along with sustainable development goals, KMU strives to contribute to the creation of innovative medical science technologies and the supply of healthcare workforce, promoting holistic societal development.
Website: https://www.kanazawa-med.ac.jp/English/public_html/
About Mr. Kousuke Kazama from Kanazawa Medical University, Japan
Mr. Kousuke Kazama is a Research Associate at the Research Support Center, Medical Research Institute, Kanazawa Medical University, Japan. His main research interests include the mechanisms of infertility and intracellular structure and epigenetics in sperm, oocyte and embryo. Mr. Kazama has over six publications in this field, with around 48 citations and over 950 reads.
Funding information
This work was supported by the Takeda Science Foundation (Grant No. 2022048848), Lotte Research Promotion Grant (Grant No. LF001353) and a Grant for Promoted Research from Kanazawa Medical University (S2023-1).
END

