Blocking a cell-to-cell tunnel slows toxic protein spread in Huntington's mice
Florida Atlantic University
Toxic mutant huntingtin protein spreads between brain cells through microscopic tube-like bridges called tunneling nanotubes, and blocking the molecular machinery that builds those tubes dramatically reduces the spread, at least in mice. That is the central finding from a team at Florida Atlantic University, published in Science Advances.
The study identifies a specific two-protein partnership that drives nanotube formation: Rhes, a protein already implicated in Huntington's disease, and SLC4A7, a bicarbonate transporter previously known only for helping cells regulate their internal pH. When the researchers eliminated SLC4A7, either genetically or with drugs, the nanotubes failed to form, and toxic protein transfer between neurons dropped sharply.
Rhes and SLC4A7: an unlikely molecular partnership
Huntington's disease is caused by an expanded CAG repeat in the huntingtin gene, producing a mutant protein that aggregates inside neurons and eventually kills them. The disease affects roughly three to seven people per 100,000 worldwide, typically appearing between ages 30 and 50. There is no cure, and current treatments manage symptoms without slowing progression. Patients typically survive 10 to 20 years after symptoms emerge.
Scientists have known for years that mutant huntingtin does not stay confined to the cells where it is produced. It spreads through the brain, particularly devastating the striatum, the region responsible for movement coordination and some cognitive functions. But the mechanism of that spread has been unclear. Proteins can move between cells via several routes: secretion into extracellular fluid, packaging into vesicles called exosomes, or direct cell-to-cell transfer.
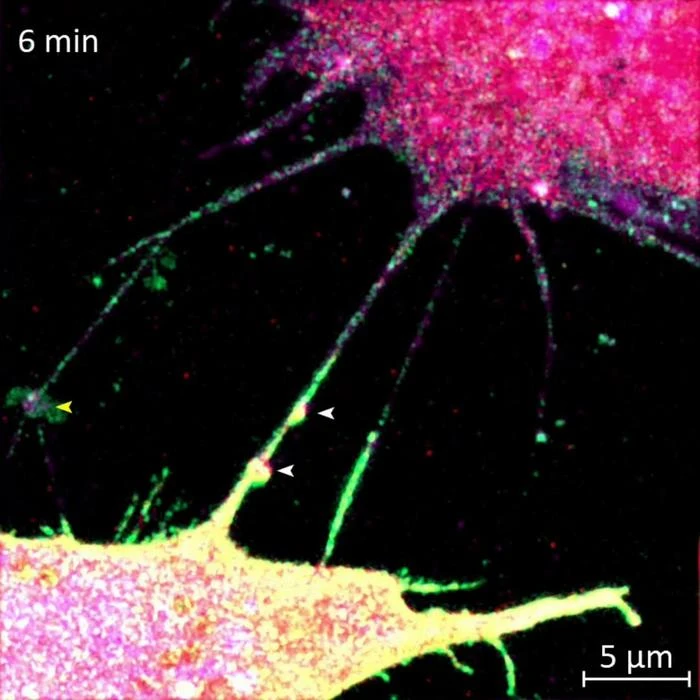
The FAU team, led by Srinivasa Subramaniam in the Department of Chemistry and Biochemistry, focused on the third option. Tunneling nanotubes are thin, membrane-enclosed bridges that physically connect two cells, allowing direct transfer of proteins, organelles, and other cargo. They have been observed in various cell types and implicated in the spread of prions, tau protein, and even drug resistance signals between cancer cells. But no one had identified the specific machinery building these tubes in the context of Huntington's disease.
Using advanced protein-mapping techniques, the researchers discovered that Rhes physically binds to SLC4A7 at the cell membrane. This binding event triggers intracellular changes that promote nanotube growth. The partnership is specific: Rhes does not build nanotubes effectively without SLC4A7, and SLC4A7 alone does not induce tube formation.
What happened when the tubes stopped forming
The team tested their finding in two ways. First, in cell cultures, they blocked SLC4A7 either by genetically knocking out the gene or by applying pharmacological inhibitors. In both cases, nanotube formation dropped, and with it, the transfer of mutant huntingtin between neurons.
Then they moved to mouse models. Mice engineered to model Huntington's disease but lacking SLC4A7 showed a dramatic reduction in toxic protein transfer between neurons in the striatum. While the paper does not provide a specific percentage reduction in the press materials, the qualitative difference was described as substantial.
This is a meaningful step because it suggests the Rhes-SLC4A7 pathway is not just one of many redundant routes for protein spread. It appears to be a major one, at least in the striatum. Blocking it had a measurable effect on disease-relevant protein transfer in a living brain.
A druggable target, but a distant drug
Subramaniam described SLC4A7 as a potentially druggable target. Membrane transporters are, as a class, among the more tractable targets for pharmaceutical intervention. They sit on the cell surface, they have well-characterized binding sites, and several existing drug classes already target related transporter families.
But several large caveats apply. SLC4A7 is not a Huntington's-specific protein. It plays a role in acid-base homeostasis across many cell types. Inhibiting it systemically could produce significant side effects by disrupting pH regulation in healthy tissues. Any therapeutic approach would need to be highly targeted, perhaps delivered directly to the striatum, or designed to interfere specifically with the Rhes-SLC4A7 interaction rather than SLC4A7 function broadly.
The study was also conducted entirely in mouse models and cell cultures. Mouse brains are smaller, structurally simpler, and recover differently from neurodegeneration than human brains. The Huntington's mouse models used in this kind of research reproduce some features of the disease, such as protein aggregation and striatal dysfunction, but not the full 10-to-20-year progression of the human condition. Whether blocking nanotube formation in mice translates to slowed disease progression in humans is unknown.
There is also a question of timing. Huntington's disease is typically diagnosed after symptoms appear, by which point significant neuronal damage has already occurred. If toxic protein has already spread widely through the striatum, blocking further nanotube-mediated transfer might slow additional damage but cannot reverse what has already happened. The therapeutic window, how early intervention must begin to make a meaningful difference, remains entirely undefined.
Beyond Huntington's: nanotubes in cancer and tauopathies
The implications of this work extend to other diseases where tunneling nanotubes play a role. Tau protein, which aggregates in Alzheimer's disease and other tauopathies, has been observed to spread via nanotubes. Cancer cells use similar structures to share survival signals, metabolic resources, and resistance mechanisms with neighboring tumor cells. If the Rhes-SLC4A7 pathway is active in these contexts as well, it could represent a common intervention point across multiple diseases.
That is a large conditional. The FAU study demonstrated the pathway's importance in Huntington's-specific models. Whether the same molecular machinery builds nanotubes in cancer cells or in neurons spreading tau has not been tested. Rhes expression varies across tissues and cell types, and its role in non-striatal neurons is less well characterized.
The mechanics of a toxic handoff
What makes this work distinctive is its specificity. Previous research established that mutant huntingtin spreads between cells. This study identifies the construction crew. Rhes provides the structural impetus; SLC4A7, sitting at the membrane, anchors and facilitates the process. The nanotube itself is not a passive channel. It is actively built by the cell, and that construction can be interrupted.
The concept of stopping disease spread by closing intercellular tunnels is conceptually appealing. Neurodegeneration in Huntington's follows a spatial pattern, spreading outward from initial sites of damage. If that spread depends on physical bridges between cells, then dismantling the bridges should contain the damage. The mouse data support this logic, though they cannot yet prove it works in humans.
For patients and families affected by Huntington's disease, a condition with a 50 percent inheritance rate from an affected parent, any new molecular target is noteworthy. SLC4A7 is not a treatment. It is not even a drug candidate yet. But it is a specific, testable hypothesis about how the disease progresses, and that is the prerequisite for everything that follows.