Training AI on quadrupole moments unlocks faster electrostatic maps for battery design
The search for better battery electrolytes is fundamentally a search for molecules with the right electrostatic personality. How a solvent molecule distributes charge across its surface determines how it interacts with lithium ions, how it behaves at electrode interfaces, and whether it will remain stable through thousands of charge cycles. The molecular electrostatic potential, or MEP, captures all of this information in a single three-dimensional map. The problem is that computing it accurately requires quantum-chemical calculations that can take days or weeks per molecule - a timeline incompatible with the high-throughput screening that modern materials discovery demands.
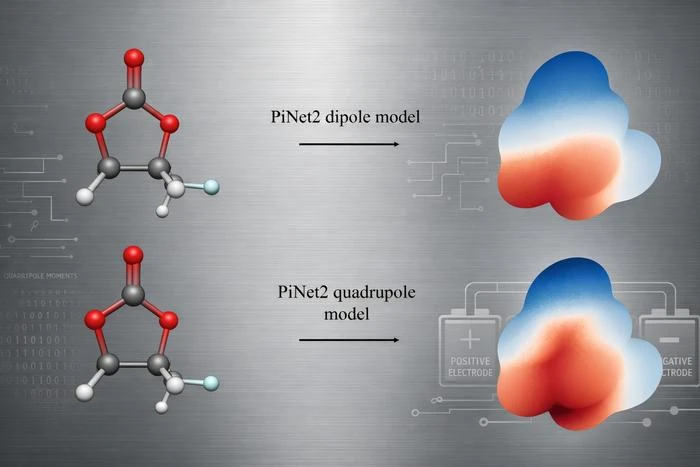
A new study from Uppsala University suggests a practical workaround: train machine learning models not just on dipole moments, as is common practice, but on quadrupole moments. The difference in prediction accuracy turns out to be substantial.
The electrostatic landscape of a solvent molecule
Every molecule generates an electrostatic field that extends into the space around it. This field governs intermolecular interactions - how the molecule binds to ions, how it organizes in solution, how it reacts at surfaces. For battery electrolytes, these interactions determine conductivity, viscosity, electrochemical stability, and ultimately device performance.
The MEP is traditionally computed using density functional theory or other quantum-mechanical methods. These calculations are accurate but computationally expensive. For a single molecule, the cost is manageable. For the thousands or tens of thousands of candidate molecules in a screening campaign, it becomes prohibitive. Machine learning models that can predict molecular properties from structural information alone offer a way to compress that computational timeline from months to hours.
Why quadrupoles carry more information than dipoles
The electrostatic potential around a neutral molecule can be decomposed into a series of terms called multipole moments. The dipole moment is the leading term - it describes the overall asymmetry of charge. The quadrupole moment is the next term, capturing how charge is distributed in a more complex, spatially detailed pattern. Think of the dipole as telling you which end of a molecule is positive and which is negative. The quadrupole tells you about the shape of that charge distribution - whether it is elongated, flattened, or twisted.
The Uppsala team, led by researcher Chao Zhang, used the PiNet2 neural network architecture to train models on both dipole and quadrupole moments, then tested how well each model could reconstruct the full electrostatic potential. They evaluated performance on two standard molecular datasets: QM9, a collection of small organic molecules commonly used for benchmarking, and SPICE, a broader and more chemically complex dataset.
The result was consistent across both datasets. Models trained on quadrupole moments produced substantially better reconstructions of the electrostatic potential than models trained on dipoles alone. The quadrupole-trained models placed effective point charges on atoms that, when combined, reproduced the spatial pattern of the full quantum-mechanical MEP with markedly higher fidelity.
Choosing the right training target
The finding carries a broader message for the growing field of machine learning in chemistry. Dipole moments have been the default training target for ML-based charge models, in part because they are the lowest-order multipole and the most widely reported in quantum-chemical databases. But this study demonstrates that being the most obvious target does not make dipoles the most informative one.
The reason is somewhat counterintuitive. When a model is trained to reproduce the dipole moment, it learns to distribute charges across atoms in a way that gets the overall charge asymmetry right. But many different charge distributions can produce the same dipole moment. The quadrupole moment adds a constraint that narrows the space of possible solutions, forcing the model to learn charge placements that are more physically realistic and that better capture the local electrostatic environment around each atom.
For battery electrolyte design, this local detail matters enormously. The interaction between a solvent molecule and a lithium ion depends on the electrostatic potential at specific locations on the molecular surface - near oxygen lone pairs, near fluorine atoms, near carbonyl groups. Getting the overall dipole right is necessary but not sufficient. The quadrupole-trained models capture these local features more accurately.
From single molecules to solvent screening
The practical application is straightforward. Battery researchers seeking new electrolyte solvents currently face a bottleneck at the screening stage. Candidate molecules must be evaluated for their ability to solvate ions, their electrochemical stability windows, and their tendency to form favorable or unfavorable solid-electrolyte interphase layers. All of these properties depend on electrostatic interactions, and all benefit from accurate MEP information.
A machine learning model that can predict reliable MEPs from molecular structure alone - without running quantum calculations for each candidate - compresses the screening timeline dramatically. The quadrupole-trained models demonstrated in this study could serve as a fast filter, identifying the most promising candidates for detailed quantum-chemical follow-up.
The energy storage field has been moving rapidly toward computational screening approaches over the past decade, driven by the sheer size of chemical space. The number of plausible electrolyte molecules runs into the millions, and experimental synthesis and testing can evaluate only a tiny fraction. Computational filters that narrow the field before lab work begins are not a luxury - they are a necessity. But those filters are only useful if their predictions are accurate enough to avoid discarding good candidates or advancing poor ones. The improved electrostatic accuracy that quadrupole training provides directly strengthens this filtering step.
Beyond batteries, the same methodology could apply to other domains where molecular electrostatics drive function: drug design, where protein-ligand binding depends on electrostatic complementarity; catalysis, where reactive intermediates are stabilized by electrostatic interactions; and materials science more broadly, where solvent selection for polymer processing or extraction chemistry depends on detailed knowledge of intermolecular forces.
Boundaries of the current work
The study has important limitations. The QM9 dataset contains only small organic molecules with up to nine heavy atoms, which are simpler than many real electrolyte solvents. The SPICE dataset is more diverse but still does not cover the full chemical space relevant to battery electrolytes, which includes fluorinated carbonates, ionic liquids, and molecules with unusual functional groups.
The models were also tested on isolated molecules in vacuum. Real electrolyte behavior involves molecules in dense liquid environments where intermolecular interactions, solvation shells, and electrode interfaces all play roles that single-molecule gas-phase calculations do not capture. Extending the approach to condensed-phase simulations remains an open challenge.
The PiNet2 architecture used here is one of many neural network frameworks for molecular property prediction. Whether the quadrupole advantage holds across other architectures - equivariant message-passing networks, transformer-based models, or Gaussian process methods - has not been tested. And the study does not validate predictions against experimental measurements of electrolyte performance; the comparison is entirely against higher-level computational benchmarks.
Still, the core finding is practical and clear: if you are building a machine learning model to predict molecular electrostatics, training on quadrupole moments gives you a better model than training on dipoles alone. For the battery electrolyte community, that represents a concrete step toward faster, more reliable computational screening of solvent candidates.